Die Bestimmung der Aminosäuren Phenylalanin und Tyrosin im Blut mittels ARACUS Aminosäureanalysator bietet eine zuverlässige Methode zur Diagnose und Überwachung der Phenylketonurie (PKU). Durch die präzise Quantifizierung und das Verhältnis dieser Aminosäuren lassen sich frühzeitig pathologische Stoffwechselveränderungen erkennen und therapeutisch begleiten. Die Methode übertrifft herkömmliche Massenspektrometrieverfahren in Genauigkeit und Differenzierungsvermögen und ist daher ideal für das Neugeborenenscreening sowie die laufende Betreuung betroffener Patienten geeignet.
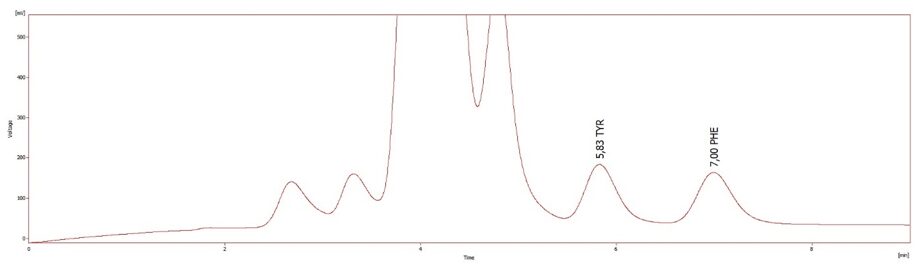
Aminosäuren Tyrosin und Phenylalanin als Biomarker für Phenylketonurie, gemessen mit dem Aminosäuranalysator ARACUS
Phenylketonurie (PKU) ist eine genetisch bedingte Stoffwechselerkrankung. Sie wird durch eine verminderte Aktivität der Phenylalanin-Hydroxylase im Körper oder einen Mangel an ihrem Coenzym Tetrahydrobiopterin verursacht, was zu einem gestörten Stoffwechsel von Phenylalanin zu Tyrosin, einem Anstieg der Phenylalaninkonzentration im Blut und in den Geweben und einem erheblichen Anstieg von Phenylpyruvat, Phenylessigsäure und Phenylmilchsäure im Urin führt. Man spricht daher von „Phenylketonurie“. Phenylalanin ist eine essentielle Aminosäure für das Wachstum und den Stoffwechsel des Menschen. Ein Teil des Phenylalanins aus der Nahrung wird für die Proteinsynthese verwendet, ein anderer Teil wird durch die Wirkung der Phenylalaninhydroxylase in Tyrosin umgewandelt, um zu funktionieren.
Symptome der Phenylketonurie
Die meisten Kinder mit PKU verhalten sich bei der Geburt normal. Während der Neugeborenenzeit treten keine besonderen klinischen Symptome auf. Unbehandelte Kinder zeigen nach 3 bis 4 Monaten allmählich geistige Retardierung und motorische Entwicklungsstörungen, ihr Haar verfärbt sich von schwarz zu gelb, die Haut wird weiß, der ganze Körper und der Urin haben einen speziellen Rattengeruch, oft haben sie Ekzeme. Mit zunehmendem Alter wird die geistige Retardierung immer deutlicher, etwa 60 % der älteren Kinder haben schwere geistige Behinderungen. Die meisten Kinder, die innerhalb eines Monats nach der Geburt behandelt werden, haben keine geistige Behinderung. Je später die Behandlung erfolgt, desto deutlicher sind die Hirnschäden.
Diagnose der Phenylketonurie
Da sich die Phenylketonurie zunächst durch einen Anstieg der Phenylalaninkonzentration im Blut bemerkbar macht, ist die Messung der Phenylalaninkonzentration im Blut die bevorzugte Methode zur Diagnose der PKU. In der Regel liegt der Phenylalaninwert im Blut bei 3120 mmol/L, was als positiv gewertet wird. Wenn gleichzeitig die Tyrosinkonzentration im Blut bestimmt werden kann, ist es für die weitere Diagnose besser, das Verhältnis von Phenylalanin zu Tyrosin zu analysieren, wobei 3/2 als positiv gewertet wird.
Behandlung der klassischen PKU, der moderaten Hyperphenylalaninämie und der moderaten bis leichten Hyperphenylalaninämie
Diese drei Typen werden durch Anomalien in der Phenylalaninhydroxylase selbst verursacht, aber es gibt kein Phenylalanin, das Medikament der Aminosäurehydroxylase, die einzige Möglichkeit, die Aufnahme von Phenylalanin aus der Nahrung zu reduzieren, ist die Kontrolle der Phenylalaninkonzentration im Körper auf ein angemessenes Niveau.
Da gewöhnliche Lebensmittel, insbesondere solche mit hohem Proteingehalt wie Fleisch, Fisch, Garnelen, Eier und Sojaprodukte, einen hohen Phenylalaningehalt aufweisen, ist es notwendig, die Aufnahme dieser Lebensmittel zu reduzieren und sie mit phenylalaninsäurefreier Milch oder Proteinpulver zu versorgen, um dem Körper andere essenzielle Aminosäuren zuzuführen. Phenylalanin ist jedoch eine der essenziellen Aminosäuren für den menschlichen Körper und muss dessen Wachstums- und Entwicklungsbedarf decken. Zu wenig Phenylalanin im Körper kann auch Krankheiten verursachen. Daher müssen diese Kinder „medizinische Spezialnahrung“ zu sich nehmen.
Das Neugeborenenscreening ist in China seit 1985 vollständig eingeführt. Es gibt drei Hauptarten von Screening und Behandlung. Die derzeitige Screening-Deckungsrate ist immer noch niedrig. Untersucht wurde das Vorkommen von PKU (Phenylketonurie) und CH (angeborene Hypothyreose). Alle untersuchten Personen wurden 72 Stunden nach der Geburt an der Ferse akupunktiert.
Im Vergleich zur traditionellen MS-Methode kann die Aminosäure-Analysemethode mehr Arten von Aminosäuren identifizieren, um den Nachteil zu beheben, dass einige Aminosäuren mit gleichen oder ähnlichen Massenspektren durch die MS-Methode nicht unterschieden werden können: zum Beispiel haben die folgenden Aminosäuren die gleiche oder ähnliche Massenspektrometrie.
- Leucin (131 17 g/mol)
- Isoleucin (131 17 g/mol)
- Allo-Isoleucin (131 17 g/mol)
- Hydroxyprolin (131 13 g/mol)
Allo-Isoleucin im Blut oder Urin ist ein signifikanter Marker für die Ahornsirup-Krankheit (MSUD).
Zu den Vorteilen der Aminosäure-Analysemethode gehören eine höhere Genauigkeit, einfache Bedienung, bessere Reproduzierbarkeit und Präzision. Die Genauigkeit ist sehr wichtig für die Überwachung des Gesundheitszustands von Patienten mit Stoffwechselstörungen. Die genaue Messung der Aminosäurekonzentration in Blutproben ist eine wichtige Grundlage für die Optimierung der Behandlung, Pflege und diätetischen Umstellung von Patienten. Durch aktive Behandlung, Pflege und Ernährungsumstellung kann die Gesundheit von Patienten mit Aminosäurestoffwechselstörungen gewährleistet werden.
Hinweis: Die zu diesem Artikel gehörige PDF ist zum Download nur auf Englisch verfügbar.